{kind=link}

We have been working from a long time ago in bivalve mollusks of comercial importance in our country (Spain), and we are also trying to find robust molecular markers to be able to differenciate autochthonous populations from alochthonous ones. In this sense, the work have been mainly cytogenetic, with remarkable results in characterizing chromosomal markers.
In addition, we are working on molecular genetics with the same objectives. After studying several species from the cytogenetic point of view, we have begun to make southern and dot blot, PCR, DNA sequencing, RAPDs, and other molecular techniques.
The lanes in which our investigation progress are: Cytogenetic characterization of several species of bivalve mollusks of comercial importance (genus Ensis, Solen, Venerupis, Ruditapes, Pecten, Mytilus, Cerastoderma, ...) from all around the world, RAPDs markers for differents populations of bivalve mollusks, characterization of ribosomal genes (spacer and coding regions), characterization of satellite DNA, microsatellite molecular markers, characterization of the mitochondrial genome and characterization of histone genes (spacer and coding regions).
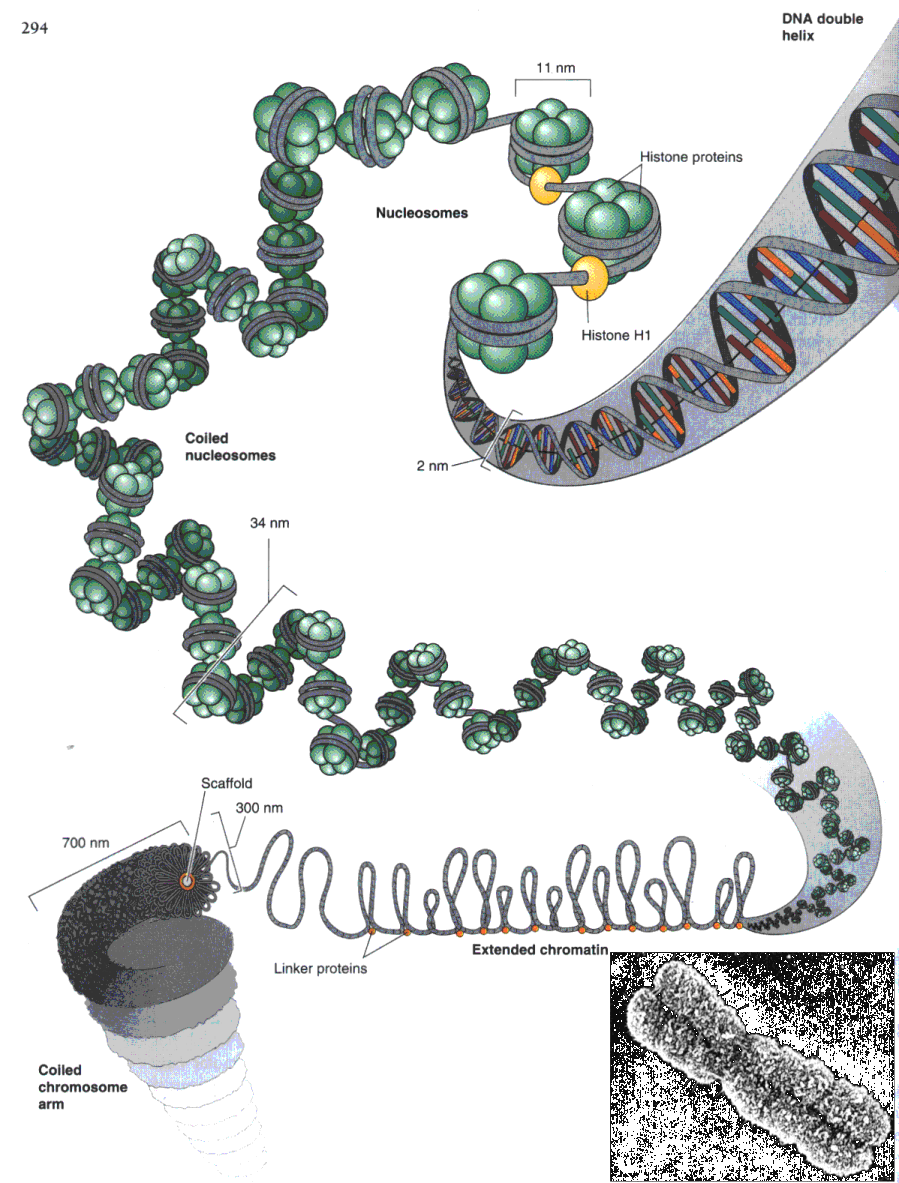
My investigation is developed in histone genes in these bivalves (get Basic Bibliographic References). Histone genes are ubiquitous in eukaryotes; their products, a family of five small, highly conserved proteins, serve to organize DNA into its fundamental unit, the nucleosome (fig. 1), and, in concert with other nucleoproteins, into higher orders of chromatin structure. Histones, particularly H3 and H4 proteins, do evolve unusually slowly, reflecting directly the very fundamental character of the functions performed by histones within the cell.
Figure 1. Model of octameric histone core based on a 3.1 Å resolution structure determined by X-ray crystalography. The histone core contains two copies each of H2A (light blue), H2B (dark blue), H3 (green), and H4 (white). The spheres represent amino acid residues, not atoms. The positively charged arginine and lysine residues are red. The amino termini of the histone proteins are not visualized by X-ray crystallography, but they are thought to extend outward from the top and bottom of this view of the histone octameric core.
The regulation of histone gene expression is known to occur at several levels. Histone mRNA levels and protein synthesis often appear coupled to the cell cycle, probably primarily due to regulation at the transcriptional level. Also, developmental regulation of histone gene expression of both a quantitative and a qualitative (variant-specific) kind also occurs, the latter type being particularly evident for the H1 class of histones during the terminal differentiation of specific tissues such as the nucleated erythroid cells of Xenopus. So, its clear that expression is regulated at several levels. A prerequisite for a greater insight into perhaps the most fundamental level of regulation - the transcriptional level - is a knowledge of the organization of the histone genes.
Histone genes are organizated in tandem array, in clusters. Generally, one or more copies of each histone gene exists into the repetitive unit (get basic information about histone genes organization, only in spanish). The more regular organization of these genes is persent in sea urchins, but this regularity turn on heterogeneity as we climb on the evolutionary scale (get general examples of histone genes organization in different organisms, only in spanish). However, microheterogeneity is allowed in invertebrates. Between coding regions, there are AT-rich spacer regions, with more variability than coding regions. These spacer regions are, at least, as long as coding regions and is hypothesized that they works as "homogeneizators" of clusters in events of unequal crossing-over. So, spacer regions mantain an uniform and regular number of genes into each repetitive unit.
In histone genes, a lack of introns seems to be characteristic of the family, although this may also be true for other gene families, notably for the interferon family, and single-copy genes as SRY. Clusters show heterogeneity, in number, organization and sense of transcription. Generally, in sea urchins, the organization is characterized by a high number of copies, with only one copy of each histone gene. Transcription direction can be divergent or convergent in adjacent genes and the repeat lenght is variable, mainly in function of spacer regions.
We have amplified by PCR up to 75% of the coding region in histone genes H1, H2B, H3 and H4 in several populations of mussels (genus Mytilus). Also, we have sequenced H1, H3 and H4 genes from PCR products. PCR primers were designed from histone genes of other organisms, and the amplification is possible because these genes are very well conserved at the nucleotide level. From PCR products, we are able to obtain good DNA probes for multiple purposes (get Basic Matherial Data).
From these probes, we can make FISH to localize histone genes on chromosomes of the species under study. We are able to perform dot-blot and in this way, we can determine the copy number of histone genes in different populations. In these data, we can search for significative differences between the populations, using them as molecular markers.
At the molecular level, automatic DNA sequencing allow us to perform multiple aligments from sequences of H1, H3, and H4 histone genes in several populations of mussels. From multiple alignments, our objective is to reconstruct gene trees for every histone, making nucleotide substitution distances (Jukes & Cantor and Tajima & Nei) and phylogenetic trees (neighbor-joining and UPGMA) from distance matrix data. From resulting gene trees, we should be able to group different mussel populations, and compare these trees (get trees inferred from molecular data in H1 and H3) between the different histone genes studied. Also, tree data inferred from histone gene sequences, will be compared with those trees inferred from RAPDs data, hierarchical cluster classifications inferred from genome-size variation data, satellite DNA analysis data, ribosomal tree data, microsatellite analysis data, chromosomal data and other types of analysis.
The integrated discussion of the data could help us to define the real species tree inside the group of mussels studied, being able to find significative differences at the molecular level between different populations, to find molecular markers in different species, and also to clarify the taxonomic status of the species under study.
![]()
| MAIN PAGE |